
Guía Esencial Para El Diseño de Electrónica Médica
La industria de dispositivos médicos creció significativamente en las últimas décadas siguiendo los avances exponenciales en electrónica, computación y ciencias de la vida. Además, el auge de la atención médica personal y móvil aumentó la demanda de tecnologías pequeñas, portátiles y de bajo consumo, lo que agregó popularidad al mercado de dispositivos médicos. Por lo tanto, no es de extrañarse que cada vez más desarrolladores de electrónica ingresen al campo. Pero diseñar electrónica médica no es una tarea fácil, y el desarrollador debe estar preparado para enfrentar desafíos y regulaciones que no se requieren en la electrónica de consumo.
Uno de los errores más comunes es no tener en cuenta las reglamentaciones pertinentes al principio del proceso de diseño, lo que lleva a modificaciones costosas o incluso al rediseño completo del dispositivo. Por ello, es importante conocer la normativa y las mejores prácticas antes de iniciar un nuevo proyecto. En este artículo, discutiremos las regulaciones y estándares más importantes para dispositivos médicos, centrándonos en los sistemas electrónicos.

Pruebas Y Estándares Para La Electrónica Médica
La regulación de los dispositivos médicos depende del país de origen: por ejemplo, la Administración de Alimentos y Medicamentos (FDA) es la principal agencia reguladora en los EE. UU., mientras que la Agencia Europea de Medicamentos (EMA) es el equivalente de la UE. Sin embargo, las recomendaciones de diseño se basan típicamente en los estándares de la Comisión Electrotécnica Internacional (IEC), en particular, IEC-60601-1 e IEC-60601-1-2. Al adherirse a estos estándares, el producto se vuelve "compatible con 601", lo cual es crucial para la aprobación de la certificación y el lanzamiento al mercado.
La IEC-60601-1 define el equipo eléctrico médico como cualquier “equipo eléctrico que tiene una parte aplicada o transfiere energía hacia o desde el paciente o detecta dicha energía hacia o desde el paciente”. En otras palabras, un dispositivo que pasa un tiempo en contacto con el usuario y/o intercambia energía con el cuerpo. Algunos ejemplos de dispositivos médicos son sensores de ECG, EEG y EMG, imágenes de ultrasonido, monitores de frecuencia cardíaca, glucómetros, termómetros digitales y marcapasos. Estos dispositivos deben pasar varias pruebas de inmunidad EMI/EMC, ESD y de fuente de alimentación para la certificación, manteniendo tanto la seguridad como el rendimiento esencial durante y después de las pruebas.
Pruebas de EMI/EMC
Estas pruebas aseguran que el dispositivo médico es compatible con el entorno electromagnético, es decir, si el dispositivo no es un generador de ruido y puede soportar la EMI de fondo sin problemas. En términos de emisión, el dispositivo debe cumplir con el Comité Especial Internacional sobre Interferencias de Radio (CISPR), en particular, el estándar CISPR 11. Hay dos tipos de EMI: la conducida y la radiada.
La EMI conducida se refiere a la energía de interferencia transmitida a través de cables, mientras que la EMI radiada corresponde a la interferencia inalámbrica. En el CISPR 11, el rango de frecuencia para las pruebas debe cubrir 150 kHz - 30 MHz (conducido) y 150 kHz - 18 GHz (radiado), con amplitudes máximas según la clase de dispositivo. Además, los armónicos de la línea eléctrica conducida deben filtrarse de acuerdo con IEC 61000-3-2, especialmente si el dispositivo utiliza fuentes de alimentación conmutadas. El ruido conducido se puede filtrar mediante perlas de ferrita, filtros de línea y estranguladores de ferrita. La regulación de EMI radiada puede cambiar de un país a otro, según la agencia reguladora.
La susceptibilidad a la radiofrecuencia es más crítica en los dispositivos médicos, ya que deben ser lo suficientemente robustos para seguir funcionando incluso en condiciones de las EMI problemáticas. Las pruebas están especificadas por IEC 60601-1-2, con un rango de frecuencia de 80 a 2700 MHz para inmunidad radiada y de 0.15 a 80 MHz para inmunidad conducida. Las amplitudes máximas varían según el dispositivo, que se puede clasificar como equipo de atención médica domiciliaria o equipo de centro médico profesional.
Durante la prueba, el dispositivo se somete a varios niveles de interferencia de radiofrecuencia en una cámara anecoica, y se analizan e informan las fallas de funcionalidad durante y después de la prueba. El dispositivo también debe ser inmune a las interferencias magnéticas de baja frecuencia (50/60 Hz) inducidas por las líneas eléctricas (IEC 61000-4-8).
Para pasar las pruebas de la EMC, tenga cuidado con el diseño del circuito y de la PCB para evitar nodos de alta impedancia y otras antenas parásitas. La implementación adecuada de filtros, compensadores de modo común, blindajes y técnicas de señalización robustas (como líneas diferenciales y cables coaxiales) puede mejorar significativamente la inmunidad de EM.
Pruebas de Inmunidad de La Fuente de Alimentación
Los dispositivos médicos conectados al sistema de distribución de energía no pueden ser demasiado susceptibles a la inestabilidad del suministro de energía, ya que pondría al paciente en un riesgo significativo. Por lo tanto, la IEC-60601 también establece varias pruebas de inmunidad, que cubren sobretensiones transitorias rápidas, parpadeo e inestabilidad de energía. El estándar IEC 61000-3-3 describe las pruebas de fluctuación de potencia y parpadeo, especificando las amplitudes máximas y los períodos de variación de voltaje que debe soportar el dispositivo durante la inspección. Para las pruebas de caídas de voltaje, se utiliza el IEC-61000-4-11.
Los dispositivos médicos deben funcionar correctamente durante y después de las caídas de tensión del 95 % durante un máximo de 5 segundos, lo que puede lograrse con baterías o bancos de condensadores. Finalmente, las normas IEC-61000-4-4 e IEC-61000-4-5 se utilizan para evaluar la susceptibilidad del dispositivo a ráfagas y sobretensiones, con diferentes longitudes y amplitudes (hasta ±2 kV).
Pruebas de Descarga Electrostática
La electrónica médica, especialmente los circuitos pequeños con contacto directo con el usuario son susceptibles a la ESD, por lo que es imprescindible una protección de la ESD robusta. El estándar para las pruebas de protección de la ESD se basa en el estándar IEC-1000-4-2. Durante la evaluación, el dispositivo bajo prueba se somete a ±8 kV en descargas de contacto y ±15 kV en descargas de aire. El dispositivo debe presentar un funcionamiento sin errores durante esta prueba, por lo que la inmunidad de la ESD básica es fundamental en cualquier diseño, pero es especialmente importante cuando se utilizan voltajes altos, como en los desfibriladores.
Matriz de Gestión de Riesgos
Una poderosa herramienta para el diseño de sistemas médicos es la matriz de gestión de riesgos. Se utiliza para proporcionar métricas de cuantificación para cada posible riesgo asociado con el dispositivo médico durante cualquier modo de operación y posible falla. Las recomendaciones internacionales para la matriz están definidas por el estándar ISO 14971:2019, que también incluye software médico y dispositivos in vitro. Aborda los riesgos relacionados con la biocompatibilidad, daños eléctricos, radiación y usabilidad, entre otros.
Se puede diseñar una matriz de gestión de riesgos mediante la distribución de la probabilidad de falla (rara, improbable, probable, esperada y cierta) en un eje y las consecuencias de la falla (menor, moderada, grave, mayor y catastrófica) en el otro eje, y llenando las celdas con el nivel de aceptación. Un ejemplo se muestra en la Tabla 1.
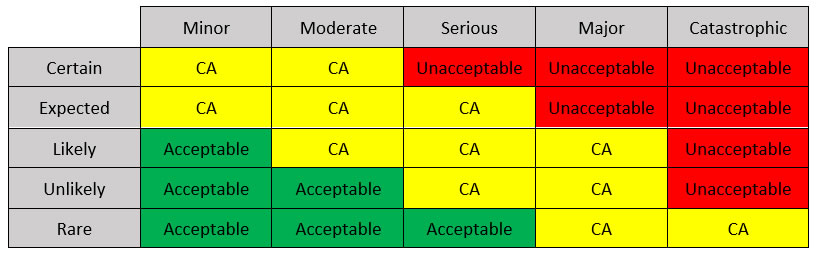
Tabla 1: Una posible Matriz de Gestión de Riesgos (CA: condicionalmente aceptable).
En la matriz de ejemplo, hay tres zonas: la región aceptable, región condicionalmente aceptable (CA) y región inaceptable. Cada uno corresponde a una combinación entre probabilidad de falla y consecuencia. Las consecuencias están relacionadas con resultados específicos de la vida real según el dispositivo y la aplicación, incluida la pérdida del sistema, daños importantes en el sistema, lesiones menores al paciente, lesiones graves al paciente y la muerte del paciente.
Los riesgos que caen en la región aceptable requieren modificaciones menores o ninguna modificación, mientras que los riesgos en la región CA requieren la identificación de mecanismos de mitigación para volverse aceptables. Los riesgos inaceptables deben ser eliminados.
Aislamiento del Paciente
Para garantizar que los problemas relacionados con los módulos electrónicos no afecten el cuerpo del paciente, es crucial proporcionar un aislamiento adecuado entre el circuito y el usuario. Es una de las partes más importantes del diseño médico, y el aislamiento inadecuado es una de las principales razones de rechazo.
El IEC-60601-1-1 establece que no puede existir una ruta de CC eléctrica entre el usuario y el dispositivo, ya que puede causar quemaduras y daños locales. Por lo tanto, cualquier lectura o inyección de señal debe realizarse mediante condensadores de desacoplamiento o de forma inalámbrica. El estándar describe reglas bien definidas para la corriente de fuga, la amplitud del voltaje, la energía y la frecuencia de la señal aplicada al paciente, así como el aislamiento dieléctrico y la separación necesaria según los niveles de voltaje. Además, cualquier parte que no esté conectada al paciente debe proporcionar buenos niveles de aislamiento, lo que se puede lograr con un espacio libre y un revestimiento adecuados.
Los transformadores de aislamiento, optoacopladores, filtros de paso alto y capacitores de desacoplamiento son algunas de las herramientas que pueden aumentar la seguridad y garantizar que el diseño pase el proceso de certificación. Además, cualquier fuente de alimentación externa, regulador o convertidor implementado debe cumplir con el estándar IEC-60601-1.
Equipo de laboratorio
Hay muchos dispositivos utilizados en la práctica médica y la investigación que no necesitan ningún contacto con el paciente, que se consideran equipos de laboratorio. En estos casos, el dispositivo debe estar diseñado de acuerdo con la norma IEC-61010, que establece los requisitos de seguridad para instrumentos y equipos eléctricos de laboratorio, y las recomendaciones de IEC-60610-1. Dispositivos como microscopios, autoclaves, analizadores químicos, equipos de citología e incubadoras de cultivos celulares son ejemplos de equipos médicos de laboratorio.

Ruta de Certificación de Electrónica Médica
Una parte integral del circuito de diseño médico es el proceso de certificación, que decide si su dispositivo está listo para comercializarse o no. Este proceso es definido, controlado y ejecutado por una agencia reguladora, que varía de un país a otro. Por ejemplo, los dispositivos médicos están regulados por la FDA en los EE. UU., por la EMA en Europa y por Health Canada en Canadá. En este artículo, nos centraremos en la regulación de la FDA, ya que el mercado médico de los EE. UU. es enorme y la FDA cumple con la mayoría de las regulaciones en todo el mundo.
Clases de Dispositivos
Las normas de la FDA para la certificación de productos dependen de la naturaleza del dispositivo médico. En este sentido, la agencia define tres clases diferentes de dispositivos para especificar a qué tipo de proceso debe someterse el producto, incluida la aplicación previa a la comercialización, la aprobación y los controles. Los dispositivos médicos se clasifican según el nivel de riesgo:
- Clase I: dispositivos que no están destinados a aplicaciones de soporte vital y/o mantenimiento de la vida, y no representan un riesgo significativo para el usuario en caso de falla. El proceso de certificación es mucho más relajado para los dispositivos de Clase I, ya que la mayoría de ellos pueden lanzarse al mercado sin ninguna notificación o aprobación previa a la comercialización. Sin embargo, todo dispositivo de Clase I debe cumplir con los controles generales de la FDA. La Clase I cubre la mayoría de los dispositivos médicos del mercado, incluidos vendajes, estetoscopios y cubrebocas quirúrgicos. Aunque la mayoría de los dispositivos de Clase I no contienen partes electrónicas críticas, los cepillos de dientes eléctricos y las camas de hospital están incluidos en la clase.
- Clase II: estos dispositivos suponen un riesgo moderado para el paciente/usuario, ya que suelen requerir largos periodos de contacto con el cuerpo y suelen tener partes invasivas. A diferencia de la mayoría de los dispositivos de Clase I, los equipos de Clase II deben pasar por el proceso de notificación previa a la comercialización, también llamado 510(k). Sin embargo, algunos dispositivos están exentos de la 510(k). La FDA también define controles especiales para regular la Clase II, que varían según la aplicación. Algunos ejemplos de los dispositivos de Clase II son sillas de ruedas eléctricas, máquinas de rayos X, bombas de infusión y lentes de contacto.
- Clase III: esta clase cubre los dispositivos médicos de alto riesgo, incluidos los equipos de soporte y mantenimiento de la vida, los implantes y cualquier equipo que presente un riesgo significativo de lesión o enfermedad para el paciente/usuario. Aproximadamente el 10% de los dispositivos médicos aprobados son de Clase III, incluidos los marcapasos, desfibriladores, implantes cocleares y ventiladores de alta frecuencia. Los controles generales y especiales no son suficientes para regular los dispositivos de Clase III, y la mayoría debe pasar por el proceso de aprobación previa a la comercialización (PMA). Los dispositivos de Clase III exentos aún deben completar la notificación 510(k) pero están libres de la PMA.
Lanzamiento Al Mercado
La FDA define varios pasos y estándares para llevar un dispositivo médico al mercado, incluidos los controles generales y específicos, la notificación previa a la comercialización: 510 (k) y la aprobación previa a la comercialización (PMA). La ruta de certificación depende de la clase y la naturaleza del dispositivo, así como de las exenciones aplicadas. Por tanto, el primer paso consiste en identificar la clase del dispositivo, lo que se puede hacer a través del sistema de paneles de clasificación desarrollado por la FDA.
Paneles de Clasificación
Una de las formas más fáciles de identificar la clase de un dispositivo es encontrar una descripción coincidente en los paneles de clasificación. La FDA dividió más de 1700 tipos de dispositivos médicos en 16 paneles (o categorías) diferentes. Cada panel está asociado con una especialidad médica, incluidos "Hospital general", "Microbiología" y "Ortopedia", que luego se dividen en tipos de dispositivos. Por ejemplo, la especialidad de “Ortopedia” cubre más de cien tipos, como “Cofia Ósea” y “Barra de Fijación Intramedular”. Cada elemento contiene una breve descripción y la clase correspondiente (I, II o III) del dispositivo. Se puede acceder a los paneles de clasificación en el sitio web de la FDA.
Controles Generales y Especiales
La FDA también establece un conjunto básico de reglas de diseño que son comunes a todos los dispositivos médicos: los controles generales. Regula, entre otros artículos, la adulteración de dispositivos, etiquetado incorrecto, registro, notificación previa a la comercialización y dispositivos restringidos. También proporciona recomendaciones generales para las mejores prácticas de fabricación. En el caso de los dispositivos de Clase I exentos, el cumplimiento de los controles generales es el único requisito antes de su lanzamiento al mercado. Sin embargo, para los dispositivos de Clase I no exentos y la mayoría de los dispositivos de Clase II, los Controles generales no son suficientes, por lo que la FDA define los controles especiales. La vigilancia posterior a la comercialización, los requisitos de datos previos a la comercialización, los requisitos especiales de etiquetado y los registros de pacientes son algunos de los temas incluidos en la sección de los controles especiales.
Notificación Previa A La Comercialización 510(K)
Además de los controles especiales, los dispositivos de clase II requieren la presentación de una notificación previa a la comercialización para la aprobación de la FDA. Este proceso a menudo se denomina 510(k), que corresponde a la sección de notificación previa a la comercialización en la Ley de Alimentos, Medicamentos y Cosméticos (FD&C). La presentación de la 510(k) es una descripción detallada del dispositivo médico, que incluye información técnica y funcional sobre la eficacia, la seguridad y los modos de funcionamiento. Para la aprobación, la FDA verifica si el dispositivo es “sustancialmente equivalente” a otro dispositivo médico que ya está en el mercado. Por lo tanto, el proceso de la 510(k) es básicamente una certificación por equivalencia. La mayoría de los dispositivos de Clase II y algunos de Clase III exentos solo necesitan enviar una notificación de la 510(k) antes del lanzamiento al mercado, por lo que buscar un equipo equivalente para evitar la necesidad de un PMA es un paso fundamental en estas clases.
Aprobación Previa A La Comercialización (PMA)
Finalmente, el último paso en el proceso de certificación de la FDA es la aprobación previa a la comercialización (PMA). El proceso de la PMA incluye una serie de ensayos clínicos y experimentos de laboratorio para certificar la efectividad y seguridad del dispositivo médico a través de los datos sólidos y consistentes. Por lo tanto, es un proceso costoso, laborioso y lento, y no cumplir con el PMA puede ser desastroso para la empresa. La FDA proporciona recomendaciones y estándares bien definidos para cada prueba, que el ingeniero puede consultar durante el proceso de diseño para evitar retiros del mercado. La mayoría de los dispositivos de Clase III se consideran equipos de riesgo elevado y no pueden comercializarse legalmente sin una certificación de la PMA, por lo que es importante identificar la clase del dispositivo e integrar los requisitos de la PMA desde el principio en el flujo de diseño.
Toallitas, Hisopos Y Solventes de Coventry Para Los Dispositivos Electrónicos Médicos Críticos
Coventry™, una marca de Chemtronics, es conocida mundialmente por resolver los desafíos de limpieza de precisión más críticos. Coventry tiene una amplia variedad de hisopos y toallitas para salas limpias para satisfacer sus aplicaciones más exigentes. El hisopo envuelto, de diseño exclusivo sin costuras ni bordes, evita que se rayen las superficies delicadas. También se incluyen en la línea de productos de tela sellada, espuma sellada y control estático.
Los Hisopos de Control Estático de la ESD de Coventry están diseñados para eliminar el daño causado por la electricidad estática. Los mangos de estos hisopos disiparán el 99 % de una carga de 5000 voltios (5 KV) en menos de 2.0 segundos. La aleación de plástico disipadora de estática utilizada para fabricar los mangos es inherentemente disipadora de estática. No contiene aditivos, revestimientos ni tensioactivos, por lo que funciona con cualquier nivel de humedad y es muy resistente a los solventes, incluso a la acetona. Elija entre nuestro poliéster lavado de Clase 10 (ISO Clase 4) o espuma de poliuretano ultralimpia de 100 poros por pulgada.
Los limpiadores solventes y los fluidos portadores de Coventry están formulados y fabricados según una especificación exacta para proporcionar un rendimiento sin igual. Tenemos productos estándar e incluso podemos diseñar soluciones personalizadas para satisfacer sus necesidades específicas.
El solvente de limpieza de precisión Coventry 12820 es una mezcla azeotrópica patentada diseñada específicamente para la limpieza y validación efectivas del hardware de los sistemas de oxígeno de propulsión de la NASA. Esta fórmula única no es combustible y exhibe buena compatibilidad con los materiales, compatibilidad con el sistema de oxígeno y alta efectividad de limpieza. Coventry 12820 es un reemplazo ideal de AK-225 en sistemas donde se requiere una alta eficiencia de limpieza y una alta eliminación de residuos no volátiles.
El Solvente Coventry™ 12809 Multi-Phase Carrier es una mezcla saturada de n-octano y perfluorocarbono (PFC). El n-octano proporciona solvencia mientras que el PFC proporciona un manto de vapor de perfluorocarbono inerte sobre la mezcla líquida. Estos vapores actúan para impedir la inflamabilidad del n-octano. A medida que el perfluorocarbono se evapora durante el uso, el PFC suplementario pasa a la solución saturada.
Chemtronics también ofrece una línea completa de solventes ultrasónicos y desengrasantes de vapor que son ideales para la electrónica médica crítica. Para obtener más información, comuníquese con su especialista en aplicaciones de Chemtronics al 678-928-6534 o [email protected].
Referencias
La FDA de los EE. UU. Dispositivos médicos, https://www.fda.gov/medical-devices, consultado en febrero del 2022
Bare, D. (2018, 21 de octubre) EMC para dispositivos médicos: EN/IEC 60601-1-2, 4.ª edición. Resúmenes de diseño médico. https://www.medicaldesignbriefs.com/component/content/article/mdb/features/articles/33060.
Klessascheck, M. (16 de marzo de 2021). IEC 60601-1-2 (Compatibilidad electromagnética de dispositivos médicos): ¡Preste atención a la nueva edición! Instituto Johnner. https://www.johner-institute.com/articles/product-development/and-more/iec-60601-1-2/.
Fenton, R. (2021, 29 de octubre). ¿Cuáles son las diferencias en las clases de dispositivos médicos de la FDA? Qualio. https://www.qualio.com/blog/fda-medical-device-classes-differences.
Haga una pregunta técnica
Manténgase actualizado sobre noticias, productos, videos y más de Chemtronics.
Productos relacionados




